



1-COORDINATION régionale et interrégionale : liens forts avec les structures de soin et de prise en charge, les équipes médicales et paramédicales, les médecins traitants, les associations, ainsi que les centres de référence ou compétences de l’inter-région.
Les Moyens :
• Réunions mensuelles de télémédecine avec les autres centres de notre inter région
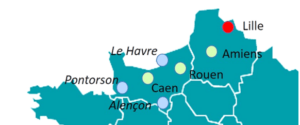
• Activité décentralisée pour apporter notre expertise au plus près des patients et de leurs familles
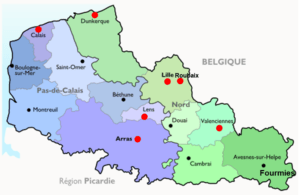
• Le CLAD Nord-Ouest fait partie de l’ERN ITHACA (label européen)
2-MEDICALE :
- Diagnostic des pathologies rares du développement et syndromes malformatifs avec ou sans déficience intellectuelle
- Coordination de la prise en charge pluridisciplinaire en lien avec le médecin traitant pour tous les patients relevant de notre centre
- Conseil génétique et prise en charge familiale (diagnostic prénatal ou préimplantatoire, diagnostic présymptomatique).
- Du fait de l’impact familial des maladies génétiques, un accompagnement psychologique est systématiquement proposé
3-EXPERTISE :
Avis d’expert syndromologiste :
- Consultations externes et au lit du patient
- Consultation multidisciplinaire :
• Coordination et participation à de multiples consultations multidisciplinaires - Participation à la commission pluridisciplinaire de diagnostic prénatal du CHU de Lille et du CH de Lens, liens étroits avec les obstétriciens et foetopathologistes
- Coordination et participation à de multiples réunions de concertation pluridisciplinaires locales, régionales et nationales, pour orienter les analyses génétiques et pour aider à leur interprétation.
4-ENSEIGNEMENT ET FORMATION / INFORMATION :
• Enseignement universitaire, postuniversitaire et en écoles paramédicales
• Liens avec les MDPH, les CAMSP et les associations de patients
• Actions grand publics ponctuelles notamment lors de la Journée Internationale Maladies Rares (JIMR)